Receptor (biochemistry)

 Clash Royale CLAN TAG#URR8PPP
Clash Royale CLAN TAG#URR8PPP 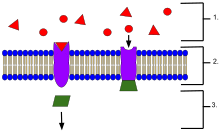
An example of membrane receptors.
- Ligands, located outside the cell
- Ligands connect to specific receptor proteins based on the shape of the active site of the protein.
- The receptor releases a messenger once the ligand has connected to the receptor.
In biochemistry and pharmacology, a receptor is a protein molecule that receives chemical signals from outside a cell.[1] When such chemical signals bind to a receptor, they cause some form of cellular/tissue response, e.g. a change in the electrical activity of a cell. There are three main ways the action of the receptor can be classified: relay of signal, amplification, or integration.[2] Relaying sends the signal onward, amplification increases the effect of a single ligand, and integration allows the signal to be incorporated into another biochemical pathway.[2] In this sense, a receptor is a protein-molecule that recognizes and responds to endogenous chemical signals, e.g. an acetylcholine receptor recognizes and responds to its endogenous ligand, acetylcholine. However, sometimes in pharmacology, the term is also used to include other proteins that are drug targets, such as enzymes, transporters, and ion channels.
Receptor proteins can be classified by their location. Transmembrane receptors include ion channel-linked (ionotropic) receptors, G protein-linked (metabotropic) hormone receptors, and enzyme-linked hormone receptors.[1] Intracellular receptors are those found inside the cell, and include cytoplasmic receptors and nuclear receptors.[1] A molecule that binds to a receptor is called a ligand, and can be a protein or peptide (short protein), or another small molecule such as a neurotransmitter, hormone, pharmaceutical drug, toxin, or parts of the outside of a virus or microbe. The endogenously designated -molecule for a particular receptor is referred to as its endogenous ligand. E.g. the endogenous ligand for the nicotinic acetylcholine receptor is acetylcholine but the receptor can also be activated by nicotine and blocked by curare.[citation needed]
Each receptor is linked to a specific cellular biochemical pathway. While numerous receptors are found in most cells, each receptor will only bind with ligands of a particular structure, much like how locks will only accept specifically shaped keys. When a ligand binds to its corresponding receptor, it activates or inhibits the receptor's associated biochemical pathway.
Contents
1 Structure
2 Binding and activation
2.1 Agonists versus antagonists
2.2 Constitutive activity
3 Theories of drug-receptor interaction
3.1 Occupation
3.2 Rate
3.3 Induced-fit
3.4 Spare Receptors
4 Receptor-regulation
5 Ligands
5.1 Extracellular
5.2 Intracellular
6 Role in genetic disorders
7 In the immune system
8 See also
9 References
10 External links
Structure
Transmembrane receptor:E=extracellular space; I=intracellular space; P=plasma membrane
The structures of receptors are very diverse and include the following major categories, among others:
- Type 1: Ligand-gated ion channels (ionotropic receptors) – These receptors are typically the targets of fast neurotransmitters such as acetylcholine (nicotinic) and GABA; and, activation of these receptors results in changes in ion movement across a membrane. They have a heteromeric structure in that each subunit consists of the extracellular ligand-binding domain and a transmembrane domain where the transmembrane domain in turn includes four transmembrane alpha helices. The ligand-binding cavities are located at the interface between the subunits.
- Type 2: G protein-coupled receptors (metabotropic receptors) – This is the largest family of receptors and includes the receptors for several hormones and slow transmitters e.g. dopamine, metabotropic glutamate. They are composed of seven transmembrane alpha helices. The loops connecting the alpha helices form extracellular and intracellular domains. The binding-site for larger peptide ligands is usually located in the extracellular domain whereas the binding site for smaller non-peptide ligands is often located between the seven alpha helices and one extracellular loop.[3] The aforementioned receptors are coupled to different intracellular effector systems via G proteins.[4]
- Type 3: Kinase-linked and related receptors (see "Receptor tyrosine kinase" and "Enzyme-linked receptor") – They are composed of an extracellular domain containing the ligand binding site and an intracellular domain, often with enzymatic-function, linked by a single transmembrane alpha helix. The insulin receptor is an example.
- Type 4: Nuclear receptors – While they are called nuclear receptors, they are actually located in the cytoplasm and migrate to the nucleus after binding with their ligands. They are composed of a C-terminal ligand-binding region, a core DNA-binding domain (DBD) and an N-terminal domain that contains the AF1(activation function 1) region. The core region has two zinc fingers that are responsible for recognizing the DNA sequences specific to this receptor. The N terminus interacts with other cellular transcription factors in a ligand-independent manner; and, depending on these interactions, it can modify the binding/activity of the receptor. Steroid and thyroid-hormone receptors are examples of such receptors.[5]
Membrane receptors may be isolated from cell membranes by complex extraction procedures using solvents, detergents, and/or affinity purification.
The structures and actions of receptors may be studied by using biophysical methods such as X-ray crystallography, NMR, circular dichroism, and dual polarisation interferometry. Computer simulations of the dynamic behavior of receptors have been used to gain understanding of their mechanisms of action.
Binding and activation
Ligand binding is an equilibrium process. Ligands bind to receptors and dissociate from them according to the law of mass action.
- [Ligand]⋅[Receptor]⇌Kd[Ligand-receptor complex](the brackets stand for concentrations)displaystyle overset underset text(the brackets stand for concentrations)[ce Ligand]cdot [ce Receptor]ce <=>[K_d][textLigand-receptor complex]
![displaystyle overset underset text(the brackets stand for concentrations)[ce Ligand]cdot [ce Receptor]ce <=>[K_d][textLigand-receptor complex]](https://wikimedia.org/api/rest_v1/media/math/render/svg/59c3c80451a744bac62346222691014b5360d93e)
One measure of how well a molecule fits a receptor is its binding affinity, which is inversely related to the dissociation constant Kd. A good fit corresponds with high affinity and low Kd. The final biological response (e.g. second messenger cascade, muscle-contraction), is only achieved after a significant number of receptors are activated.
Affinity is a measure of the tendency of a ligand to bind to its receptor. Efficacy is the measure of the bound ligand to activate its receptor.
Agonists versus antagonists
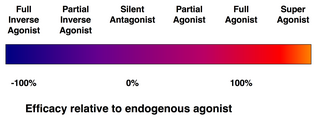
Efficacy spectrum of receptor ligands.
Not every ligand that binds to a receptor also activates that receptor. The following classes of ligands exist:
(Full) agonists are able to activate the receptor and result in a strong biological response. The natural endogenous ligand with the greatest efficacy for a given receptor is by definition a full agonist (100% efficacy).
Partial agonists do not activate receptors with maximal efficacy, even with maximal binding, causing partial responses compared to those of full agonists (efficacy between 0 and 100%).
Antagonists bind to receptors but do not activate them. This results in a receptor blockade, inhibiting the binding of agonists and inverse agonists. Receptor antagonists can be competitive (or reversible), and compete with the agonist for the receptor, or they can be irreversible antagonists that form covalent bonds (or extremely high affinity non-covalent bonds) with the receptor and completely block it. The proton pump inhibitor omeprazole is an example of an irreversible antagonist. The effects of irreversible antagonism can only be reversed by synthesis of new receptors.
Inverse agonists reduce the activity of receptors by inhibiting their constitutive activity (negative efficacy).
Allosteric modulators: They do not bind to the agonist-binding site of the receptor but instead on specific allosteric binding sites, through which they modify the effect of the agonist. For example, benzodiazepines (BZDs) bind to the BZD site on the GABAA receptor and potentiate the effect of endogenous GABA.
Note that the idea of receptor agonism and antagonism only refers to the interaction between receptors and ligands and not to their biological effects.
Constitutive activity
A receptor which is capable of producing a biological response in the absence of a bound ligand is said to display "constitutive activity".[6] The constitutive activity of a receptor may be blocked by an inverse agonist. The anti-obesity drugs rimonabant and taranabant are inverse agonists at the cannabinoid CB1 receptor and though they produced significant weight loss, both were withdrawn owing to a high incidence of depression and anxiety, which are believed to relate to the inhibition of the constitutive activity of the cannabinoid receptor.
Mutations in receptors that result in increased constitutive activity underlie some inherited diseases, such as precocious puberty (due to mutations in luteinizing hormone receptors) and hyperthyroidism (due to mutations in thyroid-stimulating hormone receptors).
Theories of drug-receptor interaction
Occupation
The central dogma of receptor pharmacology is that a drug effect is directly proportional to the number of receptors that are occupied. Furthermore, a drug effect ceases as a drug-receptor complex dissociates.
Ariëns & Stephenson introduced the terms "affinity" & "efficacy" to describe the action of ligands bound to receptors.[7][8]
Affinity: The ability of a drug to combine with a receptor to create a drug-receptor complex.
Efficacy: The ability of a drug-receptor complex to initiate a response.
Rate
In contrast to the accepted Occupation Theory, Rate Theory proposes that the activation of receptors is directly proportional to the total number of encounters of a drug with its receptors per unit time. Pharmacological activity is directly proportional to the rates of dissociation and association, not the number of receptors occupied:[9]
- Agonist: A drug with a fast association and a fast dissociation.
- Partial-agonist: A drug with an intermediate association and an intermediate dissociation.
- Antagonist: A drug with a fast association & slow dissociation
Induced-fit
As a drug approaches a receptor, the receptor alters the conformation of its binding site to produce drug—receptor complex.
Spare Receptors
In some receptor systems (e.g. acetylcholine at the neuromuscular junction in smooth muscle), agonists are able to elicit maximal response at very low levels of receptor occupancy (<1%). Thus, that system has spare receptors or a receptor reserve. This arrangement produces an economy of neurotransmitter production and release.[5]
Receptor-regulation
Cells can increase (upregulate) or decrease (downregulate) the number of receptors to a given hormone or neurotransmitter to alter their sensitivity to different molecule. This is a locally acting feedback mechanism.
- Change in the receptor conformation such that binding of the agonist does not activate the receptor. This is seen with ion channel receptors.
Uncoupling of the receptor effector molecules is seen with G-protein couple receptor.- Receptor sequestration (internalization).[10] e.g. in the case of hormone receptors.
Ligands
The ligands for receptors are as diverse as their receptors. Examples include:[11]
Extracellular
| Receptor | Ligand | Ion current |
| Nicotinic acetylcholine receptor | Acetylcholine, Nicotine | Na+, K+, Ca2+[11] |
Glycine receptor (GlyR) | Glycine, Strychnine | Cl− > HCO−3[11] |
GABA receptors: GABA-A, GABA-C | GABA | Cl− > HCO−3[11] |
Glutamate receptors: NMDA receptor, AMPA receptor, and Kainate receptor | Glutamate | Na+, K+, Ca2+[11] |
| 5-HT3 receptor | Serotonin | Na+, K+[11] |
| P2X receptors | ATP | Ca2+, Na+, Mg2+[11] |
Intracellular
| Receptor | Ligand | Ion current |
| cyclic nucleotide-gated ion channels | cGMP (vision), cAMP and cGTP (olfaction) | Na+, K+[11] |
| IP3 receptor | IP3 | Ca2+[11] |
| Intracellular ATP receptors | ATP (closes channel)[11] | K+[11] |
| Ryanodine receptor | Ca2+ | Ca2+[11] |
Role in genetic disorders
Many genetic disorders involve hereditary defects in receptor genes. Often, it is hard to determine whether the receptor is nonfunctional or the hormone is produced at decreased level; this gives rise to the "pseudo-hypo-" group of endocrine disorders, where there appears to be a decreased hormonal level while in fact it is the receptor that is not responding sufficiently to the hormone.
In the immune system
The main receptors in the immune system are pattern recognition receptors (PRRs), toll-like receptors (TLRs), killer activated and killer inhibitor receptors (KARs and KIRs), complement receptors, Fc receptors, B cell receptors and T cell receptors.[12]
See also
- Ki Database
- Ion channel linked receptors
- Neuropsychopharmacology
Schild regression for ligand receptor inhibition- Signal transduction
- Stem cell marker
- Wikipedia:MeSH D12.776#MeSH D12.776.543.750 – receptors.2C cell surface
References
^ abc Hall, JE (2016). Guyton and Hall Textbook of Medical Physiology. Philadelphia, PA: Elsevier Saunders. pp. 930–937. ISBN 978-1-4557-7005-2..mw-parser-output cite.citationfont-style:inherit.mw-parser-output qquotes:"""""""'""'".mw-parser-output code.cs1-codecolor:inherit;background:inherit;border:inherit;padding:inherit.mw-parser-output .cs1-lock-free abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-lock-subscription abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registrationcolor:#555.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration spanborder-bottom:1px dotted;cursor:help.mw-parser-output .cs1-hidden-errordisplay:none;font-size:100%.mw-parser-output .cs1-visible-errorfont-size:100%.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-formatfont-size:95%.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-leftpadding-left:0.2em.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-rightpadding-right:0.2em
^ ab Alberts B, Bray D, Hopkin K, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2014). Essential Cell Biology (Fourth ed.). New York, NY, USA: Garland Science. p. 534. ISBN 978-0-8153-4454-4.
^ Congreve M, Marshall F (March 2010). "The impact of GPCR structures on pharmacology and structure-based drug design". British Journal of Pharmacology. 159 (5): 986–96. doi:10.1111/j.1476-5381.2009.00476.x. PMC 2839258. PMID 19912230.
^ Qin K, Dong C, Wu G & Lambert NA (August 2011). "Inactive-state preassembly of G(q)-coupled receptors and G(q) heterotrimers". Nature Chemical Biology. 7 (10): 740–7. doi:10.1038/nchembio.642. PMC 3177959. PMID 21873996.
^ ab Rang HP, Dale MM, Ritter JM, Flower RJ, Henderson G (2012). Rang & Dale's Pharmacology (7th ed.). Elsevier Churchill Livingstone. ISBN 978-0-7020-3471-8.
^ Milligan G (December 2003). "Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective". Molecular Pharmacology. 64 (6): 1271–6. doi:10.1124/mol.64.6.1271. PMID 14645655.
^ Ariens EJ (September 1954). "Affinity and intrinsic activity in the theory of competitive inhibition. I. Problems and theory". Archives Internationales De Pharmacodynamie Et De Therapie. 99 (1): 32–49. PMID 13229418.
^ Stephenson RP (December 1956). "A modification of receptor theory". British Journal of Pharmacology and Chemotherapy. 11 (4): 379–93. doi:10.1111/j.1476-5381.1956.tb00006.x. PMC 1510558. PMID 13383117.
^ Silverman RB (2004). "3.2.C Theories for Drug—Receptor Interactions". The Organic Chemistry of Drug Design and Drug Action (2nd ed.). Amsterdam: Elsevier Academic Press. ISBN 0-12-643732-7.
^ Boulay G, Chrétien L, Richard DE, Guillemette G (November 1994). "Short-term desensitization of the angiotensin II receptor of bovinde adrenal glomerulosa cells corresponds to a shift from a high to low affinity state". Endocrinology. 135 (5): 2130–6. doi:10.1210/en.135.5.2130.
^ abcdefghijkl Boulpaep EL, Boron WF (2005). Medical physiology: a cellular and molecular approach. St. Louis, Mo: Elsevier Saunders. p. 90. ISBN 1-4160-2328-3.
^ Waltenbaugh C, Doan T, Melvold R, Viselli S (2008). Immunology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. p. 20. ISBN 0-7817-9543-5.
External links
- IUPHAR GPCR Database and Ion Channels Compendium
- Human plasma membrane receptome
Cell+surface+receptors at the US National Library of Medicine Medical Subject Headings (MeSH)