价层电子对互斥理论

 Clash Royale CLAN TAG#URR8PPP
Clash Royale CLAN TAG#URR8PPP 价层电子对互斥理论(英语:Valence Shell Electron Pair Repulsion,簡稱為VSEPR),是一个用来预测单个共价分子形态的化学模型。理论通过计算中心原子的价层电子数和配位数来预测分子的几何构型,并构建一个合理的路易斯结构式来表示分子中所有键和孤对电子的位置。
目录
1 理论基础
2 实际预测
3 范例
4 例外
4.1 过渡金属化合物
4.2 IIA族卤化物
4.3 一些AX2E2型分子
4.4 一些形如AX6E1的分子
5 与其他相关理论的对比
6 参考资料
理论基础
价层电子对互斥理论的基础是,分子或离子的几何构型主要决定于与中心原子相关的电子对之间的排斥作用。该电子对既可以是成键的,也可以是没有成键的(叫做孤对电子)。只有中心原子的价层电子才能够对分子的形状产生有意义的影响。
分子中电子对间的排斥的三种情况为:
- 孤对电子间的排斥(孤-孤排斥);
- 孤对电子和成键电子对之间的排斥(孤-成排斥);
- 成键电子对之间的排斥(成-成排斥)。
分子会尽力避免这些排斥来保持稳定。当排斥不能避免时,整个分子倾向于形成排斥最弱的结构(与理想形状有最小差异的方式)。
孤对电子间的排斥被认为大于孤对电子和成键电子对之间的排斥,后者又大于成键电子对之间的排斥。因此,分子更倾向于最弱的成-成排斥。
配体较多的分子中,电子对间甚至无法保持90°的夹角,因此它们的电子对更倾向于分布在多个平面上。
实际预测
下面是价层电子对互斥理论预测的分子形状表。
| 电子对数 | 杂化类型(混層類型) | 轨道形状 | 单电子对数(孤電子對) | 分子形状 | 例 |
|---|---|---|---|---|---|
| 2 | sp | 直线形 | 0 | 直线形 | BeCl2、CO2 |
| 3 | sp2 | 平面正三角形 | 0 | 平面正三角形 | BCl3 |
| 1 | V字形(角形、彎曲形) | SO2 | |||
| 4 | sp3 | 正四面体 | 0 | 正四面体 | CH4 |
| 1 | 三角锥 | NH3 | |||
| 2 | V字形(角形、彎曲形) | H2O | |||
| 5 | sp3d | 三角双锥 | 0 | 三角双锥 | PCl5 |
| 1 | 变形四面体(跷跷板形) | TeCl4 | |||
| 2 | T字形 | ClF3 | |||
| 3 | 直线形 | I3− | |||
| 6 | sp3d2 | 正八面体 | 0 | 正八面体 | SF6 |
| 1 | 四方錐 | IF5 | |||
| 2 | 平面十字形 | ICl4− | |||
| 3 | T字形 | ||||
| 4 | 直線形 | ||||
| 7 | sp3d3 | 五角雙錐 | 0 | 五角雙錐 | IF7 |
| 1 | 五角錐 | ||||
| 2 | 五角形 |
下图中,價層電子對互斥理論常用AXE方法計算分子構型。這種方法也叫ABE,其中A代表中心原子,X或B代表配位原子,E代表孤電子對。
| 电子对数 | 没有孤电子对 (基本形状) | 1个孤电子对 | 2个孤电子对 | 3个孤电子对 |
|---|---|---|---|---|
| 2 | 直线形 | | ||
| 3 |  平面三角形 |  角形 | | |
| 4 | 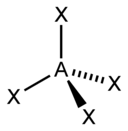 四面体形 |  三角锥形 |  角形 | |
| 5 |  三角双锥形 | 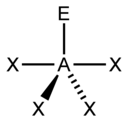 变形四面体形 |  T字形 |  直线形 |
| 6 | 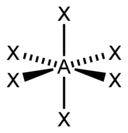 八面体形 |  四角锥形 |  平面四方形形 | |
| 7 |  五角双锥形 | 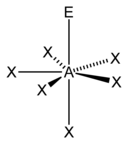 五角锥形 | 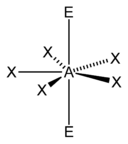 平面五角形形 | |
| 8 | 四方反棱柱形 | |
下图中:孤电子对以淡黄色球体表示。分子形状为实际几何构型,即不包含孤对电子的构型。
| 分子类型 | 分子形状 | 中心原子价电子对的排布方式† | 分子的几何构型‡ | 实例 |
|---|---|---|---|---|
| AX1En | 双原子分子 (直线形) | HF、O2 | ||
| AX2E0 | 直线形 | BeCl2、HgCl2、CO2 | ||
| AX2E1 | 角形 |  |  | NO2−、SO2、O3 |
| AX2E2 | 角形 | 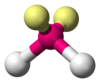 | | H2O、OF2 |
| AX2E3 | 直线形 |  | XeF2、I3− | |
| AX3E0 | 平面三角形 |  |  | BF3、CO32−、NO3−、SO3 |
| AX3E1 | 三角锥形 |  |  | NH3、PCl3 |
| AX3E2 | T字形 |  |  | ClF3、BrF3 |
| AX4E0 | 四面体形 | 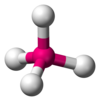 |  | CH4、PO43−、SO42−、ClO4− |
| AX4E1 | 变形四面体形 |  | 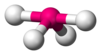 | SF4 |
| AX4E2 | 平面四方形 |  |  | XeF4 |
| AX5E0 | 三角双锥形 |  | | PCl5 |
| AX5E1 | 四角锥形 | 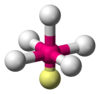 | 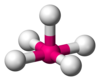 | ClF5、BrF5 |
| AX5E2 | 平面五角形 |  |  | XeF5- |
| AX6E0 | 八面体形 | 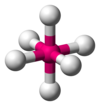 | 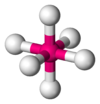 | SF6 |
| AX6E1 | 五角锥形 |  |  | XeOF5−、IOF52−[1] |
| AX7E0 | 五角双锥形 | 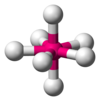 | 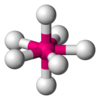 | IF7 |
| AX8E0 | 四方反棱柱形 |  |  | XeF2− 8, ZrF84-, ReF8- |
| AX9E0 | 三側錐三角柱 | 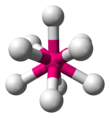 | | ReH2− 9 |
范例
甲烷分子(CH4)是四面體結構,是一個典型的AX4型分子。中心碳原子周圍有四個電子對,四個氫原子位於四面體的頂點,鍵角(H-C-H)為109°28'。
一個分子的形狀不但受配位原子影響,也受孤對電子影響。氨分子(NH3)中心原子雜化類型與甲烷相同(sp3),分子中有四個電子雲密集區,電子雲分佈依然呈四面體。其中三個是成鍵電子對,另外一個是孤對電子。雖然它沒有成鍵,但是它的排斥力影響著整個分子的形狀。因此,這是一個AX3E型分子,整個分子的形狀是三角錐形,因為孤對電子是不可「見」的。
事實上,電子對數為七是有可能的,軌道形狀是五角雙錐。但是它們僅存在於不常見的化合物之中,比如在六氟化氙中,有一對孤電子,它的構型趨向於八面體結構,因為孤對電子傾向於位於五角形的平面上。另一个例子为七氟化碘,碘沒有孤電子,七個氟原子呈五角雙錐狀排列。
電子對數為八也是有可能的,这些化合物一般为四方反棱柱体结构,[2] 例子有八氟合氙酸亚硝酰中的 [XeF8]2− 离子[3][4] 以及八氰合钼(Ⅳ)阴离子 [Mo(CN)8]4− 和八氟合锆(Ⅳ)阴离子 [ZrF8]4−。
例外
在一些化合物中VSEPR理论不能正确的预测分子空间构型。
过渡金属化合物
许多过渡金属化合物的几何构型不能用VSEPR理论解释,可以归结于价层电子中没有孤对电子以及核心的d电子与配体的相互作用。[5] 这些化合物的结构可以用VALBOND理论预测,包括金属氢化物和烷基配合物(例如六甲基钨),这个理论的基础是sd 杂化轨道和三中心四电子键模型。[6][7]晶体场理论是另一个经常可以解释配合物几何构型的理论。
IIA族卤化物
较重碱土金属的三原子卤化物的气相结构(例如:钙、锶、钡的卤化物,MX2)并不像预测的那样为直线形,而是V形。(X-M-X的大致键角: CaF2,145°;SrF2,120°;BaF2,108°;SrCl2,130°;BaCl2,115°;BaBr2,115°;BaI2,105°).[8]格莱斯皮(Ronald Gillespie)提出这是因为配体与金属原子的内层电子发生相互作用,极化使得内层电子云不是完全球面对称,因此导致了分子构型的变化。[5][9]
一些AX2E2型分子
一个例子是氧化锂分子,即Li2O,它的中间构型是直线形而不是弯曲的,这一点可以归结于如果构型是弯曲的,锂原子之间将产生强烈的排斥作用。[10]
另一个例子是O(SiH3)2(二甲硅醚)的Si-O-Si键的键角为144.1°,与其他分子中的键角相比差别较大,比如Cl2O (110.9°)、(CH3)2O (111.7°)以及N(CH3)3 (110.9°)。格莱斯皮的合理解释是孤对电子的位置不同。当配体的电负性与中心原子类似或更大时,孤对电子有能力排斥其他电子对,导致键角较小。 [5] 当中心原子电负性较大时,就像O(SiH3)2中,孤对电子的定域不明显,排斥作用较弱,这种结合导致了强配体之间的排斥(-SiH3与上面的例子相比是一个比较大的配体)使得Si-O-Si键的键角比预想的要大。[5]
一些形如AX6E1的分子
一些AX6E1型分子,例如含有Te(IV)或Bi(III)离子的化合物如TeCl62−、TeBr62−、BiCl63−、BiBr63−和BiI63−是正八面体结构;其孤对电子并不影响其构型[11]。 一种合理化解释是因为配体原子排列的拥挤没有给孤对电子留下空间[5];另一种合理化解释是惰性电子对效应。[12]
与其他相关理论的对比
价层电子对互斥理论、价键理论和分子轨道理论都是关于分子如何构成的理论。价键理论主要关注于σ键和π键的形成,通过研究受成键情况影响的轨道形状描述分子的形状。价键理论也会借助VSEPR。分子轨道理论则是关于原子和电子是如何组成分子或多原子离子的一个更精密的理论。
参考资料
^ Baran, E (2000). "Mean amplitudes of vibration of the pentagonal pyramidal XeOF5− and IOF52− anions". Journal of Fluorine Chemistry 101: 61. doi:10.1016/S0022-1139(99)00194-3.
^ Wiberg, Egon; Wiberg, Nils. Inorganic Chemistry. Arnold Frederick Holleman. Academic Press. 2001: 1165. ISBN 0123526515.
^ Peterson, Sw; Holloway, Jh; Coyle, Ba; Williams, Jm (Sep 1971). "Antiprismatic Coordination about Xenon: the Structure of Nitrosonium Octafluoroxenate(VI)". Science (New York, N.Y.) 173 (4003): 1238–1239. doi:10.1126/science.173.4003.1238. ISSN 0036-8075. PMID 17775218.
^ Hanson, Robert M. Molecular origami: precision scale models from paper. University Science Books. 1995. ISBN 093570230X.
^ 5.05.15.25.35.4 Models of molecular geometry, Gillespie R. J., Robinson E.A. Chem. Soc. Rev., 2005, 34, 396–407, doi: 10.1039/b405359c
^ Landis, C. K.; Cleveland, T.; Firman, T. K. Making sense of the shapes of simple metal hydrides. J. Am. Chem. Soc. 1995, 117, 1859–1860.
^ Landis, C. K.; Cleveland, T.; Firman, T. K. Structure of W(CH3)6. Science 1996, 272, 182–183.
^ Greenwood, N. N.; Earnshaw, A. Chemistry of the Elements 2nd. Oxford:Butterworth-Heinemann. 1997. ISBN 0-7506-3365-4.
^ Core Distortions and Geometries of the Difluorides and Dihydrides of Ca, Sr, and Ba Bytheway I, Gillespie R.J, Tang T.H., Bader R.F. Inorganic Chemistry, 34,9, 2407–2414, 1995 doi:10.1021/ic00113a023
^ A spectroscopic determination of the bond length of the LiOLi molecule: Strong ionic bonding, D. Bellert, W. H. Breckenridge, J. Chem. Phys. 114, 2871 (2001); doi:10.1063/1.1349424
^ Wells A.F. (1984) Structural Inorganic Chemistry 5th edition Oxford Science Publications ISBN 978-0-19-855370-0
^ Housecroft, C. E.; Sharpe, A. G. Inorganic Chemistry 2nd. Prentice Hall. 2004. ISBN 978-0130399137.
|