肌萎缩性脊髓侧索硬化症

 Clash Royale CLAN TAG#URR8PPP
Clash Royale CLAN TAG#URR8PPP
body.skin-minerva .mw-parser-output table.infobox captiontext-align:center
| 肌萎縮性脊髓側索硬化症 | |
|---|---|
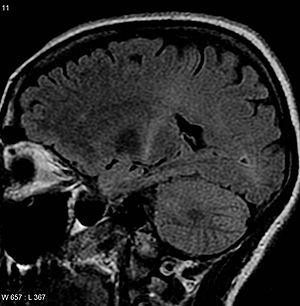 | |
肌萎缩侧索硬化症患者的核磁共振成像图。图中内囊后部(可追溯到运动皮层)出现的增强信号与肌萎缩侧索硬化症的诊断一致。 | |
| 分类和外部资源 | |
| 醫學專科 | 神經學 |
ICD-10 | G12.2 |
ICD-9-CM | 335.20 |
| OMIM | 105400 |
| DiseasesDB | 29148 |
| MedlinePlus | 000688 |
| eMedicine | neuro/14 emerg/24 pmr/10 |
| Patient UK | 肌萎缩性脊髓侧索硬化症 |
| MeSH | D000690 |
肌萎縮性脊髓側索硬化症(英语:Amyotrophic lateral sclerosis,縮寫為 ALS),也稱為肌萎缩侧索硬化症,也称为盧·賈里格症(英语:Lou Gehrig's disease)、漸凍人症、运动神经元病,是一种漸進且致命的神经退行性疾病。[1]ALS是最常見的五種運動神經元疾病(MND)之一。在英聯邦國家中,运动神经元疾病常指肌萎缩侧索硬化症。[2][3]。肌萎缩侧索硬化症由中樞神經系統內控制骨骼肌的運動神經元退化所致。由於上、下運動神經元退化和死亡,肌肉逐漸衰弱、萎縮。最後,大腦完全喪失控制隨意運動的能力。[4]最終會造成發音、吞嚥,以及呼吸上的障礙[5]。這種疾病並不一定會如阿兹海默病般影響病人的高级神经活动;相反,晚期疾病病人可一直保持清晰的思维、保留發病前的記憶、人格和智力。
肌萎缩侧索硬化症有90%至95%的发病原因不明。[4]约5%至10%遗传自父母。[6]大约有一半的病例是由两个特定基因引起的。其导致控制随意肌的神经元死亡。该病的诊断基于个人症状和体征测试,以排除其他致病的可能。[4]
肌萎缩侧索硬化症现今尚无根治方法。[4]一种名为利鲁唑的药物可以延长大约2至3月的寿命。[7]无创通气治疗可以提高患者的生活质量并延长寿命。[8]肌萎缩侧索硬化症通常在60岁左右发病,但一些直系遗传病例通常在50岁左右发病。[6]患者从发病到死亡的平均生存期为3至4年。[9]只有10%的患者生存期超过十年[4],极少数生存期为50年甚至更久。大多患者死于呼吸衰竭。世界上很多地方,肌萎缩侧索硬化症的患病率还是未知的。[6]在欧洲和美国,每年大约每十万人中就有2.2人确诊肌萎缩侧索硬化症。[6][10]
有关这种疾病的描述至少可以追溯到1824年查尔斯·贝尔的记载。[11]1869年,让-马丁·沙可首次提出了该病症状与潜在神经问题之间的联系。他在1874年开始使用“肌萎缩性侧索硬化症”这一术语。[11]棒球运动员盧·賈里格[1]及物理学家斯蒂芬·霍金罹患此病后,肌萎缩侧索硬化症才开始被人熟知。[12]2014年冰桶挑战视频在互联网上流传,提高了公众对肌萎缩侧索硬化症的认识。[13]
目录
1 症狀和體徵
1.1 早期症狀
1.2 病程
1.3 晚期
1.4 眼球运动
2 病因
2.1 遗传性
2.1.1 SOD1基因
2.2 其他因素
3 病理生理学
3.1 骨骼肌运动单元
3.2 乳酸和肉桂酸
4 诊断方法
5 处置方法
5.1 药物治疗
5.2 呼吸支持
5.3 物理治疗 / 職能治疗
5.4 营养治疗
5.5 緩和醫療
6 流行病学
7 历史
7.1 词源
8 參考文献
9 延伸閱讀
10 外部連接
症狀和體徵
由於上、下運動神經元退化導致的身體肌肉萎縮,患病者最終可能喪失發起和控制一切自主運動的能力,但膀胱、腸道和負責眼球運動的神經一般到病情晚期才会受到影響。[14]
对于大多数患者来说其认知功能并不受影响,但少数(大约5%)仍会出现额颞叶痴呆。[15]大部分患者(30–50%)出现了难以察觉的认知变化,但这一变化经过详细的神经心理学测验检测出来。极少数肌萎缩侧索硬化症患者会同时出现痴呆症、退化性肌肉疾病、退行性骨疾病,这些症状都属于多系统蛋白病综合征的一部分。[16]感觉神经和自主神经系统一般不受影响,也就意味着大多数肌萎缩侧索硬化症患者的听觉、视觉、触觉、嗅觉和味觉都能维持正常。
早期症狀
肌萎缩侧索硬化症的早期症状很难以察觉。[17]最早期的典型症狀通常是肌肉明顯的无力或/和萎缩。其他顯性的症狀包括吞嚥困難、痉挛或牵涉的肌肉僵硬。肌无力會影響四肢;或/和出現說話含糊和带鼻音的現象。肌萎缩侧索硬化症早期出现哪些症狀取決於最先受到影響的運動神經元是哪些。大約75%的患者罹患的是“四肢起病型”肌萎缩侧索硬化症,即首发症狀出現在手臂和腿部。腿起病型患者會出現走路或跑步時跌到或絆倒,走路时明显拖腿而行。臂起病型患者在做一些需要灵巧手工的事时可能会遇到困难,如扣衬衫的扣子、写字或把钥匙插入锁中。少数患者這些單手或單腳的症狀會維持較長的一段時間,被稱為單體肌萎縮。
另外约25%的患者罹患的是“延髓起病型”肌萎缩侧索硬化症,即以說話障礙或吞嚥困難为首要表现。說話開始變得含糊、帶有鼻音或失语。其他症狀包括吞嚥困難和舌頭喪失灵活性。很少一部分患者属于“呼吸起病型”肌萎缩侧索硬化症,即支持呼吸的肋間肌最先受到影响。也很少一部分患者可能伴随出现額葉癡呆症,並最終發展為其他更典型的症状。
隨著病情的發展,病人開始出現運動、吞嚥和構音障礙。上運動神經元受影響的症狀包括肌肉僵硬和痙攣、誇張的反射(反射亢進)以及過度的嘔吐反射。一個被稱為「巴宾斯基征」的異常反射也是上運動神經元受損的表現。下運動神經元退化的症狀包括肌肉无力和萎縮,可见的短暂的皮下肌肉抽搐(颤动)。大約15-45%的病人經歷假性延髓症狀——一種被稱為「情緒不穩」的神經系統疾病,症狀包括無法控制的大笑、哭泣或微笑。一個人只有在同時出現上下運動神經受損的症狀而又沒有其他的病因時,才应被診斷為肌萎缩侧索硬化症。
病程
雖然發病的順序和程度因人而异,但是大部分病人最終都會無法行走或使用他們的雙手和手臂。他們也會喪失說話和吞嚥食物的能力,並最終需要依靠被稱為BiPAP的便攜式呼吸機。病情發展的程度可以通過《肌萎缩侧索硬化症功能評定量表修訂版(ALSFRS-R)》來衡量。該測量方式是一個包含12個項目的臨床訪談或自我報告的問卷調查,评分為0分(重度殘疾)至48分(功能正常)。雖然有極少病人的發病過程緩慢,但是平均而言,病人每個月會喪失大約0.9的分值。一份對醫師的調查表明,他們認為20%的分值減少才會有臨床意義。[18]不管哪部分機體首先受到影響,肌肉的无力和萎縮都會隨著病情的發展而蔓延到其他肢体。腿部起病的患者通常會從先發病的一隻腿蔓延到另一隻腿,而延髓起病的患者通常病症會先蔓延到手臂,再到腿。
40歲以下[19][20]、輕度肥胖[21]、並只有一肢起病、主要有上運動神經元症狀.[22]的病人的病程會比較慢。相反,延髓起病、呼吸系統起病或前額葉癡呆起病的病人病程發展較快[22]。
CX3CR1等位基因突变也會影響病人病程和存活的時間。[23]
晚期
雖然呼吸机可以緩解呼吸問題並延長生存期,但是卻不會減緩肌萎缩侧索硬化症的病程。大部分肌萎缩侧索硬化症患者從病發開始3至5年内死於呼吸衰竭。從發病到死亡的中位生存時間为39個月。只有約4%的病人存活超過10年。[24]吉他手傑森·貝克於1989年發病至今,而物理學家史蒂芬·霍金存活了55年[25],他們都是十分罕見的病例。
咀嚼和吞嚥困難都會增加進食的難度,以及窒息和將食物吸入肺部的風險。在患病的晚期,吸入性肺炎會加重,而保持正常的體重也會越加困難,並有可能需要通過插管進食。隨著膈肌和肋間肌的无力,呼吸也會開始衰弱。肺功能指標,比如肺活量和吸氣壓力都會下降。呼吸起病患者,這些症狀可能在四肢發病前出現。
在患病的最後期,控制眼球運動的動眼神經以及眼外肌也會被影響。眼球運動直到最後期才受到影響很大一個原因是骨骼肌與眼外肌的差异。最終,病人的情況可能類似閉鎖綜合症。[26]
眼球运动
肌萎缩侧索硬化症患者可能会难以进行自发、快速的眼球移动。眼动速度会减慢,并且平滑追踪眼动和收敛性眼动也会出现问题。[27]通过检测前庭眼反射可以帮助识别这些问题。眼电图(EOG)技术可以测量视网膜的静息电位。EOG结果显示患者表现出了与疾病进展相关的渐进的变化,并为疾病进展对眼部运动的影响提供了临床评价度量标准。此外,EOG也许可用于眼睛问题的早期检测。[28]
眼外肌的胚细胞系不同于体节衍生的肌肉的胚细胞系。眼外肌很独特,因为它们在人的一生中会持续不断地重塑并保持个体随着年龄增长也能一直拥有活跃的卫星细胞。眼外肌明显有着比四肢骨骼肌更多的生肌前体细胞。[29]
病因
遗传性
大約5%-10%的肌萎缩侧索硬化症來自家族遺傳。[6]总而言之,第一近亲中存在一个肌萎缩侧索硬化症患者,患病的风险则增加1%。[30][31]
21號染色體上的一個超氧化物歧化酶基因突变與20%家族遺傳性肌萎缩侧索硬化症相關,佔全部病例的2%。[32][33][34]這個一突变通過常染色体显性遗传方式傳遞的,并有一百多種不同的突變类型。引發肌萎缩侧索硬化症的最常見突變是SOD1基因突變。多見於北美患者;其特征是从发病到死亡的病程极快。在斯堪的納維亞國家中最常见的突变是D90A-SOD1突变,其所造成的肌萎缩侧索硬化症病程較慢。且患者的存活时间平均可达11年。[35]
2011年,一種六核苷酸重複序列的基因異常被發現於名為C9orf72的區域。這種突變被認為與肌萎缩侧索硬化症-額葉癡呆症肌萎(ALS-FTD)有聯繫。[36]6%的歐洲白人病例与此突变相關。[37]這個基因出现于菲律賓的後裔中。[37]
UBQLN2基因在细胞中编码泛醌蛋白2,该蛋白是泛醌蛋白家族中的一种并能控制泛素化蛋白的降解。UBQLN2突变可阻碍蛋白质降解,导致神经退行性疾病,伴X染色体显性遗传肌萎缩侧索硬化症及肌萎缩侧索硬化症/痴呆。[38]
一些基因突变已被证实与多种类型的肌萎缩侧索硬化症相关。已知的联系有:
| 类型 | OMIM | 基因 | 基因座 | 备注 |
|---|---|---|---|---|
| ALS1 | 105400 | SOD1 | 21q22.1 | 最常见的肌萎缩侧索硬化症形式 |
| ALS2 | 205100 | ALS2 | 2q33.1 | |
| ALS3 | 606640 | ? | 18q21 | |
| ALS4 | 602433 | SETX | 9q34.13 | |
| ALS5 | 602099 | ? | 15q15.1–q21.1 | 青少年发病 |
| ALS6 | 608030 | FUS | 16p11.2 | |
| ALS7 | 608031 | ? | 20p13 | |
| ALS8 | 608627 | VAPB | 20q13.3 | |
| ALS9 | 611895 | ANG | 14q11.2 | |
| ALS10 | 612069 | TARDBP | 1p36.2 | |
| ALS11 | 612577 | FIG4 | 6q21 | |
| ALS12 | 613435 | OPTN | 10p13 | |
| ALS13 | 183090 | ATXN2 | 12q24.12 | |
| ALS14 | 613954 | VCP | 9p13.3 | 与肌萎缩侧索硬化症强相关[16][39] |
| ALS15 | 300857 | UBQLN2 | Xp11.23–p11.1 | 描述自一个家庭中[40] |
| ALS16 | 614373 | SIGMAR1 | 9p13.3 | 青少年发病,非常罕见,描述自一个家庭中[41] |
| ALS17 | 614696 | CHMP2B | 3p11 | 非常罕见,报告自少数患者 |
| ALS18 | 614808 | PFN1 | 17p13.3 | 非常罕见,描述自少数中国家庭[42] |
| ALS19 | 615515 | ERBB4 | 2q34 | 非常罕见,描述自2013年底的四个人[43] |
| ALS20 | 615426 | HNRNPA1 | 12q13 | 非常罕见,描述自2013年底的四个人[44] |
| ALS-FTD | 105550 | C9orf72 | 9p21.2 | 6%欧洲白人的致病原因 |
SOD1基因
在1993年,科学家们发现基因(SOD1)突变所生产的铜/锌超氧化物歧化酶(SOD1)与约20%的家族性肌萎缩性脊髓侧索硬化症相关。该酶是一种很强的抗氧化剂,能保护机体免受来源于线粒体的自由基损伤。自由基是细胞在正常代谢过程中产生的高度活性分子。自由基可积聚并导致细胞内DNA和蛋白质的损伤。到目前为止,超过110种不同的SOD1突变已被发现与代谢失调相关,其中一些(如H46R)有一个很长的临床过程;而另一些则异常迅猛,如A4V。若氧化应激防御失败,细胞则会走向凋亡。
SOD1缺陷可能是一种功能性缺失或增添。SOD1功能性缺失可能导致自由基积累。SOD1功能性增添则可能在其他方面作为毒物。[45][46]
利用转基因小鼠研究SOD1在SOD1变异导致的家族性肌萎缩侧索硬化症中所扮演的角色方面已经取得了一些理论成果。SOD1基因完全缺失的小鼠通常不会罹患肌萎缩侧索硬化症,尽管会表现出与年龄有关的肌肉萎缩加速和寿命缩短。这表明SOD1变异产生的毒蛋白会导致功能的增益而不是缺失。另外,蛋白质聚集已被发现是家族性和散发性肌萎缩侧索硬化症的共同病理特征。有趣的是,存在SOD1突变的小鼠(最常见的G93A突变),突变SOD1的聚集体(错误折叠蛋白的积累)只出现于病变组织中,更多检测于运动神经元的变性过程中[47]。突变SOD1的总积累被怀疑会通过破坏线粒体、蛋白酶体、蛋白质折叠的分子伴侣或其他蛋白质而引起细胞功能受损[48]。以上任何一个环节如果被证实,将大大增加聚集体涉及突变SOD1毒性理论的可信度。批评人士指出,在人体中由SOD1突变导致的病例仅仅占所有病例的2%左右,并且发病机制可能不同于这些少数情况。到目前为止,ALS-SOD1小鼠仍然为临床前研究该疾病的最好模型,但人们也希望能开发更多有用的模型。
已存为科学界和公众提供有关肌萎缩侧索硬化症遗传学最新信息的在线数据库ALSOD。这一网站最初是在1999年为SOD1基因而开发的,但逐渐升级为包括了超过40种与肌萎缩侧索硬化症相关的基因。
其他因素
目前大约90%的病例没有家族病史,即不能明确病因。尚不确定的潜在病因包括头部外伤,兵役,频繁用药和参与身体接触性运动。
研究还集中于谷氨酸在运动神经元变性中的作用。谷氨酸是大脑中的一种神经递质。科学家发现与健康人相比肌萎缩侧索硬化症患者的血清和脑脊液中的谷氨酸含量更高[33]。利鲁唑是目前唯一的经FDA批准用于治疗肌萎缩侧索硬化症及目标谷氨酸运载体的药物。它对患者生存只有少量的的影响,然而研究表明过量的谷氨酸并不是导致疾病的唯一原因。
某些研究表明在少数ALS病例中,尤其是运动员,其患病与富含支链氨基酸的饮食,常见导致细胞过度兴奋的膳食补充剂之间存在联系。所提出的潜在机制是细胞过度兴奋导致了细胞对钙的吸收增加,进而导致了对钙缓冲能力极差的神经细胞死亡。[49][50]
一些证据支持超氧化物歧化酶1(SOD1)蛋白在错误折叠传播上类似于朊病毒。[51]同样,也有人提出蓝藻毒素β-甲基-1-丙氨酸(BMAA)的结合导致了其他脘状蛋白质错误折叠的传播。[52][53]
另一个非常常见的病因是运动神经元系统病变,诸如额颞叶。[54]这些地方的病变经常在早期就可表现出功能缺损的迹象,这一点可用来预测运动功能丧失的方向,同时这也是造成了肌萎缩侧索硬化症恶化的原因。 肌萎缩侧索硬化症的病理变化在出现任何明显的迹象或症状前就已存在。[54][55]在病程中,大约三分之一的运动神经元在肌肉萎缩变得明显之前就已死亡。[55]
对其他一些潜在原因,包括化学品接触,电磁场暴露,职业因素,身体创伤和电击,也已进行了调查,但并未达成一致的结论。[56]
病理生理学
肌萎缩侧索硬化症的典型特征是在大脑皮层,脑干和脊髓的上下运动神经元的死亡。在死亡之前,运动神经元在胞体和轴突中会产生富含蛋白质的包裹体。这可能是由于蛋白质降解缺陷造成的。[57]这些包裹体通常含有泛素,并且通常包括与肌萎缩侧索硬化症相关的蛋白中的一种,可能是SOD1,TAR DNA结合蛋白(TDP-43或TARDBP),或/和FUS。[58]
骨骼肌运动单元
尽管共享复原的固定序列,眼外肌(EOMs)仍和骨骼肌表现出不同的特点。以下是眼外肌不同于骨骼肌运动单位的特性。[59]
- 一条神经纤维只与一条或两条肌纤维连接
- 尽管肌梭丰富但无眼的牵张反射
- 无反回性抑制
- 无特殊的快肌或慢肌
- 所有的眼部运动神经元同等的参与所有类型的眼球运动——没有专门用于扫视或平滑追踪的神经元
正常的眼外肌和患者的眼外肌之间也发现存在差异。来自尸体捐赠者的眼外肌相比于四肢肌肉能保存其细胞结构。[60]正常的眼外肌由面向眼球的中心整体层(GL)和面向眼睑壁的薄眼睑层(OL)组成。[60]受肌萎缩侧索硬化症影响的眼外肌也能保存GL和OL组织。 眼外肌具有神经营养因子——脑源性神经营养因子(BDNF)和胶质细胞源性神经营养因子(GDNF),这些神经营养因子能保存受肌萎缩侧索硬化症影响的眼外肌。[60]层粘连蛋白是一种通常存在于神经肌肉接头(NMJ)的结构蛋白。Lnα4一种是层粘连蛋白亚型,是骨骼肌神经肌肉接头的标志。[61] 肌萎缩侧索硬化症患者显示能保持眼外肌神经肌肉接头Lnα4的表达,但这种表达在此人的肢体肌肉神经肌肉接头中是不存在的。[61]层粘连蛋白的表达可能在保护肌萎缩侧索硬化症患者眼外肌的完整性中起了作用。散发性肌萎缩侧索硬化症(sALS)患者细胞内钙水平增加,导致神经递质释放的增加。散发性肌萎缩侧索硬化症患者体内血清的被动转移增加了脊髓中自发递质的释放,但不是眼外肌终端[62];因此,眼外肌被假定能耐受肌萎缩侧索硬化症导致的生理环境变化。
然而,研究者也注意到了一些现象。相比于同龄的健康者,肌萎缩侧索硬化症患者的眼外肌纤维尺寸更大。眼外肌的病变特征表现为集中和分散萎缩以及纤维肥厚,但眼外肌损伤与来自同一供体的四肢肌肉相比显著偏低。这些眼外肌还表现为在脂肪和结缔组织增生以补偿失去及萎缩的肌肉纤维。[60]在肌萎缩侧索硬化症患者中也出现了眼肌麻痹,在眼球运动核中以及附近的神经元损失。[27]此外,眼外肌纤维的肌球蛋白重链成分也发生了变化,表现为中心整体层正常表达的MyHCslow缺失,薄眼睑层不包含通常表达的MyHCemb。这种变化也许代表神经支配模式的变化,可能包括不同类型的运动神经元出现神经再支配,或缺失多个神经支配。眼外肌仅发生为MyHCslow和MyHCemb的变化,使得眼外肌纤维的成分相对正常。因为眼外肌通常是高度支配,任何去神经支配都可由邻近轴突补偿。[60]
乳酸和肉桂酸
乳酸是糖酵解的最终产品,可导致肌肉疲劳。乳酸脱氢酶(LDH)可发挥双重功能,并能够将乳酸氧化为丙酮酸,从而被三羧酸循环利用。在眼外肌中,乳酸通路能够在活动量增加的情况下能维持肌肉收缩。因此,眼外肌中高活性的乳酸脱氢酶被认为可抵抗肌萎缩侧索硬化症。[63]
肉桂酸是乳酸运输和摄入以耐疲劳性的外源乳酸的抑制剂。肉桂酸能够引起眼外肌疲劳,同时减少眼外肌耐力和残余力量;然而,肉桂酸对腿部的趾伸长肌没有影响。相比之下,以外源乳酸代替葡萄糖代谢增加了趾伸长肌的疲劳状况,而非减轻。[63]眼外肌疲劳仅仅出现于外源乳酸和肉桂酸共同取代葡萄糖代谢时。[63] Fatiguability in EOMs was only found when a combination of exogenous lactacte plus cinnamate replaced glucose.[63]
诊断方法

临床诊断患肌萎缩侧索硬化症者核磁共振(轴向液体衰减反转恢复序列)显示内囊后部T2信号增强
虽然单一肢体的上、下运动神经元症状强烈暗示患肌萎缩侧索硬化症的可能,但是仍没有测验能为肌萎缩侧索硬化症提供确切的诊断。相反,肌萎缩侧索硬化症的诊断主要根据医生对个体临床症状和体征的观察及一系列测试以排除其他疾病的可能。医生需要获得病人的全部病史,并通常经过定期的神经系统检测,评估如肌肉无力,肌肉萎缩,反射亢进,痉挛等症状是否恶化。[4]
因为肌萎缩侧索硬化症的症状可能类似于大量其他更可治愈的疾病或症状,所以必须进行适当的测试以排除患其他疾病的可能。其中一项测试是肌电图(EMG),一种检测肌肉电活动的特殊记录技术。某些肌电图结果可支持肌萎缩侧索硬化症的诊断。另一项常见的测试测量神经传导速度(NCV)。但是神经传导速度结果异常也可能是由于周围神经病变(周围神经损伤)或肌病(肌肉疾病)而不是肌萎缩侧索硬化症。肌萎缩侧索硬化症常进行磁共振成像(MRI)检查,以排除脊髓肿瘤、多发性硬化症、颈部椎间盘突出、脊髓空洞症及颈椎病。[4]
除了根据病人的症状和测试结果进行判断外,医生还可以进行血液和尿液样本检测以排除其他疾病的可能性。在某些情况下,如果医生怀疑病人可能有肌病而不是肌萎缩侧索硬化症可进行肌肉活检。[4]
某些病毒性疾病可能会导致与肌萎缩侧索硬化症类似的症状,如人类免疫缺陷病毒(HIV)、人类T细胞白血病病毒(HTLV)、莱姆病[64]、梅毒[65]和蜱传脑炎病毒[66]。神经系统疾病的某些方面也与其类似,如多发性硬化症、脊髓灰质炎后综合征、多灶性运动神经病、慢性炎症性脱髓鞘性多发性神经病、脊髓性肌萎缩症和脊髓延髓肌萎缩症。[4]
肌萎缩侧索硬化症必须与“肌萎缩侧索硬化症类似综合症”区分开,这是一种临床表现和特征与肌萎缩侧索硬化症或其变体类似的无关疾病。[67]由于其症状与早期肌萎缩侧索硬化症相似,所以患者应听取神经学专家的意见,从而排除一些临床可能。
但是,大多数肌萎缩侧索硬化症病例很容易诊断,大型肌萎缩侧索硬化症诊所的误诊率小于10%。[68][69]在一项研究中,190位患者符合MND / ALS的诊断标准按照研究规程和定期监测进行实验。三十例患者(16%)在临床观察发展阶段得出了与之前完全不同的诊断。[70]在同一项研究中,三名患者出现假阴性的诊断——重症肌无力(MG),一种自身免疫性疾病。重症肌无力症状类似于肌萎缩侧索硬化症和其他神经系统疾病,所以有时会导致延误诊断和治疗。重症肌无力是可以治疗的;肌萎缩侧索硬化症却不能。[71]肌无力综合征,也被称为伊顿-兰伯特综合征,类似于肌萎缩侧索硬化症并且其初期症状与肌萎缩侧索硬化症相似。[72][73]
处置方法
肌萎缩侧索硬化症的处置以缓解症状与延长寿命为目的。该支持治疗最好由多学科的专业医护人员进行,尽可能的保持患者可运动及舒适。
药物治疗
利鲁唑是唯一可略微提高肌萎缩侧索硬化症患者生存率的药物。[74]其可延长数月生存期,并可能对延髓起病型患者延长生存期更有效。利鲁唑还可以推迟患者恶化至需要机械通气支持。服用利鲁唑的患者必须监测肝损害(发生于约10%的用药患者)。[75]利鲁唑由美国食品药品监督管理局批准,并由英国国家卫生与临床优化研究所推荐。但利鲁唑不能修复已有的运动神经元损伤。[76]
其他药物可用来帮助减轻疲劳,缓解肌肉痉挛,控制痉挛状态,减少过多的唾液和痰。药物也可以帮助患者缓解疼痛,抑郁,睡眠障碍,吞咽困难,便秘等症状。巴氯芬和安定(地西泮)常被用来控制肌萎缩侧索硬化症引起的痉挛,当肌萎缩侧索硬化症患者出现吞咽口水困难时,可用苯海索或阿米替林。[14]
呼吸支持
当协助呼吸的肌肉逐渐衰弱,可通过协助通气(间歇正压通气,双水平正压通气(BiPAP),或双相胸甲通气(BCV))辅助呼吸。这些装置通过这些直接应用于面部或身体可人为地增加患者的肺功能。当肌肉无力维持氧气和二氧化碳的水平时,这些设备就必须全天使用。双相胸甲通气可通过高频振荡及积极的呼气性呼吸协助,有增加清除呼吸道分泌物的优势。[77]人们最终可能会考虑机械通气的方式(呼吸器),即使用机器帮助肺收缩和膨胀的方式。为了使得效果更加理想,可能需要从鼻或口连接一个管子至气管。若要长期使用,这可考虑通过氣管切開術,用一个塑料呼吸管通过颈部的开口直接插在病人的气管上。
当患者及家属决定是否及何时使用以上某一方法时,应该考虑以下几个方面。通气设备对患者生活质量的作用及所需费用因人而异。尽管呼吸支持可以缓解患者的呼吸问题并延长生存期,但它并不能阻止肌萎缩侧索硬化症病程。在作出进行通气支持的决定前,患者需充分了解这些因素以及不能移动对生活造成的长期影响,并应对生活质量问题进行深入讨论。
运用双水平正压通气的外部通气设备常被用于支持呼吸,最初用于晚上,之后也用于白天。 双水平正压通气的使用(更多时候被称为无创通气,NIV)只是一个暂时性治疗措施,在BPAP失效前,患者需决定是否进行气管切开术并长期机械通气。关于这一点,一些患者选择了姑息治疗。
物理治疗 / 職能治疗
物理治疗在肌萎缩侧索硬化症患者的病情改善上起到了很大作用。具体来说,物理及职业治疗师可以设定康复目标,并通过延长力量衰退的时间,维持耐力,减缓疼痛,预防并发症的发生,促进功能自理能力等方面提升肌萎缩侧索硬化症患者的身体素质。[78]
職能治疗和特殊设备等辅助技术也可以提高患者在肌萎缩侧索硬化症进程中的自理和安全。温和的低强度有氧运动,如日常生活自理活動、散步、游泳、骑自行车都可以锻炼未受影响的肌肉,改善心血管健康,并可帮助患者对抗疲劳和憂鬱。由職能治療師設計的功能性运动和關節伸展可以帮助患者预防痛苦的痉挛和肌肉的缩短(挛缩)。职能治疗师可協助病患在肌肉無超過负荷的情况下得到锻炼。他们建议使用如坡道、吊带、代步车、卫浴设备(淋浴椅,廁所扶手等)和轮椅来帮助患者轉位移动。职能治疗师可以提供或推荐设备并且可调整使人们能安全地自理日常生活。
说话困难的肌萎缩侧索硬化症患者或许能够在语言病理学家那里获得帮助。这些专业保健人员可以教病人自适应策略,如一些能帮助他们说话更响亮、更清楚的方法。随着肌萎缩侧索硬化症恶化,语音病理学家会推荐使用一些强化和替代性交流的设备,如扩声器,语音发生装置(或语音输出通信设备)和/或低技术含量的通信技术,如字母板或是/否的信号。
营养治疗
肌萎缩侧索硬化症患者患者和护理人员可以向营养师学习如何计划和准备大量的小餐,保证每天可提供足够的热量,纤维和流质食物以及如何改善吞咽困难。患者可能已经开始使用吸痰器来吸除多余的液体或唾液以防止窒息。职业治疗师可以协助推荐一些适合的设备使患者自我进食更容易些。语言病理学家可推荐一些食物,以迎合他们特殊的缺陷和能力。当患者不能通过自主进食而摄取足够营养时,医生可以建议使用胃管。使用胃管可以减少因将液体吸入肺部而引起窒息和肺炎的风险。进食管不会带来疼痛,也不会妨碍病人自主进食。
研究人员表示,“肌萎缩侧索硬化症患者长期能量摄入不足,建议增加能量摄入”[79]并伴有严重的食欲不振[80]。有关动物[81]表明,应鼓励肌萎缩侧索硬化症患者消耗尽可能多的热量,且不要限制他们的热量摄入。截至2012年,对于体重减轻的管理依旧存在"干预措施缺乏坚实的诊据"的问题。[82]
緩和醫療
社会工作者、家庭护理和临终关怀护士会帮助肌萎缩侧索硬化症患者,他们的家人和照顾者应对医疗、情感、经济方面的问题,特别是在疾病的晚期。社会工作者可提供,如经济援助、安排律师,准备遗嘱、为病人和医护人员的寻找支持团体等方面提供支持。家庭护士不仅可以提供医疗保健,而且会教导照顾者如何使用呼吸器、喂食,及在移动病人时避免皮肤疼痛问题和挛缩。家庭临终关怀护士会咨询医生,以确保适当的药物治疗,控制疼痛,和其他会提高希望留在家里的患者的生活质量。家庭临终关怀团队也对病人及照顾者就临终问题提供专业咨询。
流行病学
在世界上许多地区,肌萎缩侧索硬化症的发病率都还是未知的。[6]在欧洲,每年每十万人中约有2.2人确诊。[6]在美国,每年有超过5600人确诊,有超过3万患者。肌萎缩侧索硬化症每年约导致十万中2人死亡。[83]
虽然肌萎缩侧索硬化症被归入罕见疾病,但其却是最常见的运动神经元疾病。所有种族及民族背景都可能患病。每年每十万人中就有一到二人罹患肌萎缩性侧索硬化症。[84]据估计每十万白种人中有1.2至4.0人罹患肌萎缩性侧索硬化症,其他种族患病率则较低。[85]菲律宾人肌萎缩性侧索硬化症发病率仅次于白人,为每十万人中有1.1至2.8人患病。[55]
有一些关于“集群”患病的报道,包括旧金山49人队的三名橄榄球运动员[86],意大利的超过50个足协球员[87],英国南部的足协球员的三个朋友,以及法国南部的一对配偶(丈夫和妻子)。[88][89][90][91][92]尽管一些人认为肌萎缩性侧索硬化症是基因和环境共同作用的结果,但除了随年龄增长风险更高外,仍未确认环境对该病的影响。
历史
肌萎缩性侧索硬化症至少可以追溯至1824年查尔斯·贝尔的描述。[11]
英国科学家奥古斯都·沃勒在1850年描述了萎缩神经纤维的外观。1869年,法国医生让-马丁·沙可首次将这些症状与神经系统病变联系在一起,并在1874年发表论文以肌萎缩性侧索硬化症为名介绍此病。[11]1881年,该论文被翻译成了英语,出版在《神经系统疾病讲座》(Lectures on the Diseases of the Nervous System)第三卷。
美国传奇棒球运动员盧·賈里格在1939年发生罹患此病,于7月4日发表了告别演说,肌萎缩性侧索硬化症就此在美国而闻名。两年后盧·賈里格即离世。[93]2014年,因冰桶挑战而在全世界闻名。[94]
在20世纪50年代,一次肌萎缩性侧索硬化症疫情于关岛的查莫罗人中爆发。
1991年,研究者发现了21号染色体与家族性肌萎缩性侧索硬化症(FALS)联系在一起。1993年,发现21号染色体的SOD1基因在某些家族性肌萎缩性侧索硬化症病例中发挥的作用。1996年,利鲁唑成为第一个美国食品药品监督局批准用于肌萎缩性侧索硬化症的药物。
1998年,埃斯科里亚尔标准(El Escorial)被确定为肌萎缩性侧索硬化症患者临床研究分类的标准。第二年,修订后的肌萎缩性侧索硬化症功能评分量表发表,并很快成为肌萎缩性侧索硬化症患者临床研究恶化评级的黄金标准。2011年,发现C9ORF72中非编码的重复序列扩增是导致肌萎缩性侧索硬化症和额颞叶痴呆的主要原因。
词源
肌萎缩性侧索硬化症的学名来源于希腊文“amyotrophia”:其中“a-”表示“不”,“myo”指“肌肉”,trophia意思是“营养”;因此“amyotrophia”表示“没有肌肉营养”,描述了患者肌肉组织的萎缩特征。脊髓横向识别领域是受影响的神经细胞所在部位。在这一地区的退化会导致疤痕或硬化。
參考文献
^ 1.01.1 Kelly, Evelyn B. Encyclopedia of human genetics and disease. Santa Barbara, Calif.: Greenwood. 2013: 79–80. ISBN 978-0-313-38713-5.
^ Motor neurone disease. http://www.nhs.uk/. [2 January 2015].
^ Ellison, edited by Seth Love, David N. Louis, David W. Greenfield's neuropathology 8th ed. London: Hodder Arnold. 2008: 947. ISBN 978-0-340-90681-1. 引文格式1维护:冗余文本 (link)
^ 4.04.14.24.34.44.54.64.74.8 Motor Neuron Diseases Fact Sheet. National Institute of Neurological Disorders and Stroke (NINDS). [7 November 2010]. (原始内容存档于2014年4月13日).
^ Motor Neuron Diseases Fact Sheet. National Institute of Neurological Disorders and Stroke (NINDS). [7 November 2010]. (原始内容存档于2014年4月13日).
^ 6.06.16.26.36.46.56.6 Kiernan, MC; Vucic, S; Cheah, BC; Turner, MR; Eisen, A; Hardiman, O; Burrell, JR; Zoing, MC. Amyotrophic lateral sclerosis.. Lancet. 12 March 2011, 377 (9769): 942–55. PMID 21296405. doi:10.1016/s0140-6736(10)61156-7.
^ Miller, RG; Mitchell, JD; Moore, DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND).. The Cochrane database of systematic reviews. 14 March 2012, 3: CD001447. PMID 22419278. doi:10.1002/14651858.CD001447.pub3.
^ McDermott, CJ; Shaw, PJ. Diagnosis and management of motor neurone disease.. BMJ (Clinical research ed.). 22 March 2008, 336 (7645): 658–62. PMID 18356234. doi:10.1136/bmj.39493.511759.be.
^ Malamut, edited by Joseph I. Sirven, Barbara L. Clinical neurology of the older adult 2nd ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. 2008: 421. ISBN 978-0-7817-6947-1. 引文格式1维护:冗余文本 (link)
^ Epidemiology of Sporadic ALS. http://aces.stanford.edu/. [2 January 2015]. (原始内容存档于2015年10月8日).
^ 11.011.111.211.3 Rowland LP. How amyotrophic lateral sclerosis got its name: the clinical-pathologic genius of Jean-Martin Charcot. Arch. Neurol. March 2001, 58 (3): 512–5. PMID 11255459. doi:10.1001/archneur.58.3.512.
^ Youngson, David B. Jacoby, Robert M. Encyclopedia of family health 3rd ed. Tarrytown, NY: Marshall Cavendish. 2004: 1256. ISBN 978-0-7614-7486-9. 引文格式1维护:冗余文本 (link)
^ Song, P. The Ice Bucket Challenge: The public sector should get ready to promptly promote the sustained development of a system of medical care for and research into rare diseases.. Intractable & rare diseases research. August 2014, 3 (3): 94–6. PMID 25364651. doi:10.5582/irdr.2014.01015.
^ 14.014.1 Dugdale DC, Hoch DB, Zieve D. Amyotrophic lateral sclerosis. A.D.A.M. Medical Encyclopedia. 27 August 2010.
^ Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007, 6 (11): 994–1003. PMID 17945153. doi:10.1016/S1474-4422(07)70265-X.
^ 16.016.1 Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurrò MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, Galassi G, Scholz SW, Taylor JP, Restagno G, Chiò A, Traynor BJ. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron. 2010, 68 (5): 857–864. PMC 3032425. PMID 21145000. doi:10.1016/j.neuron.2010.11.036.
^ Amyotrophic Lateral Sclerosis (ALS) Fact Sheet. [2015-01-02]. (原始内容存档于2015-01-04).
^ Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler (Journal Article). 2010, 11 (1–2): 178–80. PMID 19634063. doi:10.3109/17482960903093710.
^ Sabatelli M, Madia F, Conte A, Luigetti M, Zollino M, Mancuso I, Lo Monaco M, Lippi G, Tonali P. Natural history of young-adult amyotrophic lateral sclerosis. Neurology. 16 September 2008, 71 (12): 876–81. PMID 18596241. doi:10.1212/01.wnl.0000312378.94737.45.
^ [Survival in amyotrophic lateral s... [Srp Arh Celok Lek. 1997 Jan-Feb] - PubMed – NCBI
^ Mild Obesity Appears to Improve Survival in ALS Patients. [2015-06-26]. (原始内容存档于2015-06-26). 已忽略文本“ HMS ” (帮助)
^ 22.022.1 Chiò A, Calvo A, Moglia C, Mazzini L, Mora G. Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. Journal of Neurology, Neurosurgery & Psychiatry. 2011, 82 (7): 740–746. PMID 21402743. doi:10.1136/jnnp.2010.235952.
^ Lopez-Lopez A, Gamez J, Syriani E, Morales M, Salvado M, Rodríguez MJ, Mahy N, Vidal-Taboada JM. CX3CR1 Is a Modifying Gene of Survival and Progression in Amyotrophic Lateral Sclerosis. PLoS ONE. 2014, 9 (5): e96528. PMC 4013026. PMID 24806473. doi:10.1371/journal.pone.0096528.
^ Turner MR, Parton MJ, Shaw CE, Leigh PN, Al-Chalabi A. Prolonged survival in motor neuron disease: a descriptive study of the King's database 1990–2002. J Neurol Neurosurg Psychiatry. 2003, 74 (7): 995–997. PMC 1738535. PMID 12810805. doi:10.1136/jnnp.74.7.995.
^ Stephen Hawking serves as role model for ALS patients. CNN. 2009-04-20. [永久失效連結]
^ James L. Bernat: Ethical Issues in Neurology, p. 336 http://books.google.pl/books?id=o732GWGjPQ4C&pg=PA336
^ 27.027.1 Cohen B, Caroscio J. Eye movements in amyotrophic lateral sclerosis. J Neural Transm Suppl. 1983;19:305-15
^ Palmowski A, Jost WH, Prudlo J, Osterhage J, Käsmann B, Schimrigk K, Ruprecht KW. Eye movement in amyotrophic lateral sclerosis: a longitudinal study. Ger J Ophthalmol. 1995, 4 (6): 355–62. PMID 8751101.
^ Kallestad KM, Hebert SL, McDonald AA, Daniel ML, Cu SR, McLoon LK. Sparing of extraocular muscle in aging and muscular dystrophies: a myogenic precursor cell hypothesis. Exp. Cell Res. 2011, 317 (6): 873–85. PMC 3072110. PMID 21277300. doi:10.1016/j.yexcr.2011.01.018.
^ Cookson, Mark R.; Wingo, Thomas S.; Cutler, David J.; Yarab, Nicole; Kelly, Crystal M.; Glass, Jonathan D. The Heritability of Amyotrophic Lateral Sclerosis in a Clinically Ascertained United States Research Registry. PLoS ONE. 2011, 6 (11): e27985. ISSN 1932-6203. doi:10.1371/journal.pone.0027985.
^ Sontheimer, Harald. Diseases of the Nervous System. Academic Press. 2015: 170 [2015-05-02]. ISBN 978-0-12-800403-6.
^ Conwit RA. Preventing familial ALS: A clinical trial may be feasible but is an efficacy trial warranted?. Journal of the Neurological Sciences. December 2006, 251 (1–2): 1–2. ISSN 0022-510X. PMID 17070848. doi:10.1016/j.jns.2006.07.009.
^ 33.033.1 Al-Chalabi A, Leigh PN. Recent advances in amyotrophic lateral sclerosis. Current Opinion in Neurology. August 2000, 13 (4): 397–405. ISSN 1473-6551. PMID 10970056. doi:10.1097/00019052-200008000-00006.
^ Battistini S, Ricci C, Lotti EM, Benigni M, Gagliardi S, Zucco R, Bondavalli M, Marcello N, Ceroni M, Cereda C. Severe familial ALS with a novel exon 4 mutation (L106F) in the SOD1 gene. Journal of the Neurological Sciences. June 2010, 293 (1): 112–115. PMID 20385392. doi:10.1016/j.jns.2010.03.009.
^ Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keränen ML, Bergmark L, Saarinen A, Haltia T, Tarvainen I, Kinnunen E, Udd B, Marklund SL. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation, A clinical and genealogical study of 36 patients. Brain. 1996, 119 (4): 1153–1172. PMID 8813280. doi:10.1093/brain/119.4.1153.
^ DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS (PDF). Neuron. 2011, 72 (2): 245–56. PMC 3202986. PMID 21944778. doi:10.1016/j.neuron.2011.09.011. (原始内容 (PDF)存档于2014-11-29).
^ 37.037.1 Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012, 11 (4): 323–30. PMC 3322422. PMID 22406228. doi:10.1016/S1474-4422(12)70043-1.
^ Han-Xiang Deng, Wenjie Chen, Seong-Tshool Hong 等. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 8 September 2011, 477 (7363): 211–215. PMC 3169705. PMID 21857683. doi:10.1038/nature10353. 引文格式1维护:显式使用等标签 (link)
^ Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 20 June 2013, 153 (7): 1461–74. PMC 3760148. PMID 23791177. doi:10.1016/j.cell.2013.05.037.
^ Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011, 477 (7363): 211–215. PMC 3169705. PMID 21857683. doi:10.1038/nature10353.
^ Al-Saif A, Al-Mohanna F, Bohlega S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Annals of Neurology. 2011, 70 (6): 913–919. PMID 21842496. doi:10.1002/ana.22534.
^ Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, Kost JE, Gonzalez-Perez P, Fox AD, Adams J, Taroni F, Tiloca C, Leclerc AL, Chafe SC, Mangroo D, Moore MJ, Zitzewitz JA, Xu ZS, van den Berg LH, Glass JD, Siciliano G, Cirulli ET, Goldstein DB, Salachas F, Meininger V, Rossoll W, Ratti A, Gellera C, Bosco DA, Bassell GJ, Silani V, Drory VE, Brown RH, Landers JE. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012, 488 (7412): 499–503. PMC 3575525. PMID 22801503. doi:10.1038/nature11280.
^ Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, Belzil VV, Dion PA, Higasa K, Doi K, Ishiura H, Mitsui J, Date H, Ahsan B, Matsukawa T, Ichikawa Y, Moritoyo T, Ikoma M, Hashimoto T, Kimura F, Murayama S, Onodera O, Nishizawa M, Yoshida M, Atsuta N, Sobue G, Fifita JA, Williams KL, Blair IP, Nicholson GA, Gonzalez-Perez P, Brown RH, Nomoto M, Elenius K, Rouleau GA, Fujiyama A, Morishita S, Goto J, Tsuji S. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am. J. Hum. Genet. 2013, 93 (5): 900–5. PMC 3824132. PMID 24119685. doi:10.1016/j.ajhg.2013.09.008.
^ Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in the prion-like domains of hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 28 March 2013, 495 (7442): 467–73. PMC 3756911. PMID 23455423. doi:10.1038/nature11922.
^ Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998, 281 (5384): 1851–4. PMID 9743498. doi:10.1126/science.281.5384.1851.
^ Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996, 13 (1): 43–7. PMID 8673102. doi:10.1038/ng0596-43.
^ Furukawa Y, Fu R, Deng HX, Siddique T, O'Halloran TV. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci USA. 2006, 103 (18): 7148–53. PMC 1447524. PMID 16636274. doi:10.1073/pnas.0602048103.
^ Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006, 52 (1): 39–59. PMID 17015226. doi:10.1016/j.neuron.2006.09.018.
^ Manuel M, Heckman CJ. Stronger is not always better: could a bodybuilding dietary supplement lead to ALS?. Exp Neurol (Review). March 2011, 228 (1): 5–8. PMC 3049458. PMID 21167830. doi:10.1016/j.expneurol.2010.12.007.
^ Carunchio I, Curcio L, Pieri M, Pica F, Caioli S, Viscomi MT, Molinari M, Canu N, Bernardi G, Zona C. Increased levels of p70S6 phosphorylation in the G93A mouse model of amyotrophic lateral sclerosis and in valine-exposed cortical neurons in culture. Experimental Neurology. 2010, 226 (1): 218–230. PMID 20832409. doi:10.1016/j.expneurol.2010.08.033.
^ Fernández-Borges N, Eraña H, Elezgarai SR, Harrathi C, Gayosso M, Castilla J. Infectivity versus seeding in neurodegenerative diseases sharing a prion-like mechanism. Int J Cell Biol. 25 September 2013, 2013: 583498. PMC 3800648. PMID 24187553. doi:10.1155/2013/583498.
^ Holtcamp, W. The emerging science of BMAA: do cyanobacteria contribute to neurodegenerative disease?. Environmental health perspective. 2012, 120 (3). PMC 3295368. PMID 22382274. doi:10.1289/ehp.120-a110.
^ Rodgers KJ. Non-protein amino acids and neurodegeneration: The enemy within. Experimental Neurology. March 2014, 253: 192–196. PMID 24374297. doi:10.1016/j.expneurol.2013.12.010.
^ 54.054.1 Rosenbohm, A., Kassubek, J., Weydt, P., Marroquin, N., Volk, A., Kubisch, C., Huppertz, H., & Weber, M. (2014). Can lesions to the motor cortex induce amyotrophic lateral sclerosis? . J Neurol, 261, 283-290.
^ 55.055.155.2 Walling A. Amyotrophic lateral sclerosis: Lou Gehrig's disease. American Family Physician. 1999, 59 (6): 1489–96.
^ Sutedja NA, Fischer K, Veldink JH, van der Heijden GJ, Kromhout H, Heederik D, Huisman MH, Wokke JJ, van den Berg LH. What we truly know about occupation as a risk factor for ALS: a critical and systematic review. Amyotrophic Lateral Sclerosis. 2009, 10 (5–6): 295–301. PMID 19922116. doi:10.3109/17482960802430799.
^ Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult onset ALS and ALS/dementia. Nature. 2011-08-21, 477 (7363): 211–5. PMC 3169705. PMID 21857683. doi:10.1038/nature10353.
^ Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, Mishra M, Ajroud-Driss S, Heller S, Sufit R, Siddique N, Mugnaini E, Siddique T. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. June 2010, 67 (6): 739–48. PMID 20517935. doi:10.1002/ana.22051.
^ Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science. McGraw-Hill; 2000
^ 60.060.160.260.360.4 Ahmadi M, Liu JX, Brännström T, Andersen PM, Stål P, Pedrosa-Domellöf F. Human extraocular muscles in ALS. Invest Ophthalmol Vis Sci. 2010;51(7):3494-501
^ 61.061.1 Liu JX, Brännström T, Andersen PM, Pedrosa-Domellöf F. Different Impact of ALS on Laminin Isoforms in Human Extraocular Muscles Versus Limb Muscles. Invest Ophthalmol Vis Sci. 2011
^ Mosier DR, Siklós L, Appel SH. Resistance of extraocular motoneuron terminals to effects of amyotrophic lateral sclerosis sera. Neruology. 2000;54(1):252-5
^ 63.063.163.263.3 Andrade FH, McMullen CA. Lactate is a metabolic substrate that sustains extraocular muscle function. Pflugers Arch. 2006, 452 (1): 102–8. PMID 16328456. doi:10.1007/s00424-005-0010-0.
^ Hänsel Y, Ackerl M, Stanek G. ALS-like sequelae in chronic neuroborreliosis. Wien Med Wochenschr. 1995, 145 (7–8): 186–8. PMID 7610670.
^ el Alaoui-Faris M, Medejel A, al Zemmouri K, Yahyaoui M, Chkili T. Amyotrophic lateral sclerosis syndrome of syphilitic origin. 5 cases. Rev Neurol (Paris). 1990, 146 (1): 41–4. PMID 2408129.
^ Umanekiĭ KG, Dekonenko EP. Structure of progressive forms of tick-borne encephalitis. Zh Nevropatol Psikhiatr Im S S Korsakova. 1983, 83 (8): 1173–9. PMID 6414202.
^ Silani V, Messina S, Poletti B, Morelli C, Doretti A, Ticozzi N, Maderna L. The diagnosis of Amyotrophic lateral sclerosis in 2010. Archives italiennes de biologie. 2011, 149 (1): 5–27. PMID 21412713. doi:10.4449/aib.v149i1.1260.
^ Eisen, A. Amyotrophic lateral sclerosis: A review. BCMJ. 2002, 44 (7): 362–366.
^ Davenport RJ, Swingler RJ, Chancellor AM, Warlow CP. Avoiding false positive diagnoses of motor neuron disease: lessons from the Scottish Motor Neuron Disease Register. J. Neurol. Neurosurg. Psychiatr. 1996, 60 (2): 147–51. PMC 1073793. PMID 8708642. doi:10.1136/jnnp.60.2.147.
^ Chieia MA, Oliveira AS, Silva HC, Gabbai AA. Amyotrophic lateral sclerosis: Considerations on diagnostic criteria (PDF). Arquivos de Neuro-Psiquiatria. 2010, 68 (6): 837–842. PMID 21243238. doi:10.1590/S0004-282X2010000600002.
^ Al-Asmi A, Nandhagopal R, Jacob PC, Gujjar A. Misdiagnosis of Myasthenia Gravis and Subsequent Clinical Implication: A case report and review of literature. Sultan Qaboos University medical journal. 2012, 12 (1): 103–108. PMC 3286704. PMID 22375266. doi:10.12816/0003095.
^ Lambert-Eaton Myasthenic Syndrome (LEMS). Misc.medscape.com. [18 April 2013].
^ LEMS.com, Lambert-Eaton Myasthenic Syndrome: About. Lems.com. [18 April 2013].
^ Carlesi C, Pasquali L, Piazza S, Lo Gerfo A, Caldarazzo Ienco E, Alessi R, Fornai F, Siciliano G. Strategies for clinical approach to neurodegeneration in Amyotrophic lateral sclerosis. Archives italiennes de biologie. March 2011, 149 (1): 151–67. PMID 21412722. doi:10.4449/aib.v149i1.1267.
^ Miller RG, Mitchell JD, Lyon M, Moore DH. Miller, Robert G, 编. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database of Systematic Reviews. 2007, (1): CD001447. PMID 17253460. doi:10.1002/14651858.CD001447.pub2.
^ Russell P, Harrison R. What is amyotrophic lateral sclerosis. Clinical Pharmacist. 2014, 6 (7).
^ Sviri S, Linton DM, van Heerden PV. Non-invasive Mechanical Ventilation Enhances Patient Autonomy in Decision-Making Regarding Chronic Ventilation. Critical Care and Resuscitation. Jun 2005, 7 (2): 116–118. PMID 16548804.
^ Lewis M, Rushanan S. The role of physical therapy and occupational therapy in the treatment of Amyotrophic Lateral Sclerosis. NeuroRehabilitation. 2007, 22 (6): 451–461. PMID 18198431.
^ Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ. Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am J Clin Nutr. Jan 1996, 63 (1): 130–7. PMID 8604660. doi:10.1093/ajcn/63.1.130.
^ Holm T, Maier A, Wicks P, Lang D, Linke P, Münch C, Steinfurth L, Meyer R, Meyer T. Severe Loss of Appetite in Amyotrophic Lateral Sclerosis Patients: Online Self-Assessment Study. Interact J Med Res. Apr 2013, 2 (1): e8. PMC 3632382. PMID 23608722. doi:10.2196/ijmr.2463.
^ Hamadeh MJ, Rodriguez MC, Kaczor JJ, Tarnopolsky MA. Caloric restriction transiently improves motor performance but hastens clinical onset of disease in the Cu/Zn-superoxide dismutase mutant G93A mouse. Muscle Nerve. Feb 2005, 31 (2): 214–20. PMID 15625688. doi:10.1002/mus.20255.
^ Payne, C; Wiffen, PJ; Martin, S. Interventions for fatigue and weight loss in adults with advanced progressive illness.. The Cochrane database of systematic reviews. 18 January 2012, 1: CD008427. PMID 22258985. doi:10.1002/14651858.cd008427.pub2.
^ ALS Association. Quick Facts About ALS & The ALS Association.
^ ALS Topic Overview. [2008-05-01]. (原始内容存档于2008年5月1日).
^ Gordon, Paul. "Amyotrophic Lateral Sclerosis. Pathophysiology, Diagnosis and Management." CNS Drugs, 25.1 (2011):1-15.
^ Sla, indagini nei club. Pesticidi nel mirino. [2008-10-02]. (原始内容存档于2008年10月3日).
^ Wicks P, Abrahams S, Masi D, Hejda-Forde S, Leigh PN, Goldstein LH. The Prevalence of Depression and Anxiety in MND. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2005, 6 (Supplement 1): 147. ISSN 1466-0822.
^ Rachele MG, Mascia V, Tacconi P, Dessi N, Marrosu F, Giagheddu M. Conjugal amyotrophic lateral sclerosis: a report on a couple from Sardinia, Italy. Ital J Neurol Sci. April 1998, 19 (2): 97–100. PMID 10935845. doi:10.1007/BF02427565.
^ Poloni M, Micheli A, Facchetti D, Mai R, Ceriani F, Cattalini C. Conjugal amyotrophic lateral sclerosis: toxic clustering or change?. Ital J Neurol Sci. April 1997, 18 (2): 109–12. PMID 9239532. doi:10.1007/BF01999572.
^ Camu W, Cadilhac J, Billiard M. Conjugal amyotrophic lateral sclerosis: a report on two couples from southern France. Neurology. March 1994, 44 (3 Pt 1): 547–8. PMID 8145930. doi:10.1212/WNL.44.3_Part_1.547.
^ Cornblath DR, Kurland LT, Boylan KB, Morrison L, Radhakrishnan K, Montgomery M. Conjugal amyotrophic lateral sclerosis: report of a young married couple. Neurology. November 1993, 43 (11): 2378–80. PMID 8232960. doi:10.1212/WNL.43.11.2378.
^ Corcia P, Jafari-Schluep HF, Lardillier D, Mazyad H, Giraud P, Clavelou P, Pouget J, Camu W. A clustering of conjugal amyotrophic lateral sclerosis in southeastern France. Neurol. November 2003, 60 (4): 553–7. PMID 12707069. doi:10.1001/archneur.60.4.553.
^ Farewell Speech. lougehrig.com. 4 July 1939 [16 April 2008]. (原始内容存档于12 April 2008). 已忽略未知参数|df=(帮助)
^ George Bush delivers possibly the best ALS ice bucket challenge yet. Independent. [20 August 2014].
延伸閱讀
.mw-parser-output .refbeginfont-size:90%;margin-bottom:0.5em.mw-parser-output .refbegin-hanging-indents>ullist-style-type:none;margin-left:0.mw-parser-output .refbegin-hanging-indents>ul>li,.mw-parser-output .refbegin-hanging-indents>dl>ddmargin-left:0;padding-left:3.2em;text-indent:-3.2em;list-style:none.mw-parser-output .refbegin-100font-size:100%
ALS Hope Foundation. [2008-06-21]. (原始内容存档于2008年5月12日).Dedicated to the care and cure of people with Lou Gehrig's Disease. (from site page us.php)
Lou Gehrig: The Official Web Site. CMG Worldwide. [2008-06-21]. (原始内容存档于2008年5月9日).The Official Web site of Lou Gehrig is an informational Web site intended to honor the life, the legend and the career of Lou Gehrig. (from site page siteinfo/index.htm)
Patrick Aebischer; Ann C. Kato. Playing defense against Lou Gehrig's Disease (Paper). Scientific American (Verlagsgruppe Georg von Holtzbrinck). November 2007: 86–93 [2008-06-21].Researchers have proposed potential therapies for a paralyzing disorder once thought to be untreatable (sub-title)
引文使用过时参数coauthors (帮助)[永久失效連結]- Stephen Hawking's voice in 2017
外部連接
| 分類 |
|
|---|---|
| 外部資源 |
|
维基共享资源中相关的多媒体资源:肌萎缩性脊髓侧索硬化症 |
开放式目录计划中和肌萎缩性脊髓侧索硬化症相关的内容
Template:Amyotrophic lateral sclerosis
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||